Related articles
Secondary antibodies resources
Alexa Fluor secondary antibodies
Biotinylated secondary antibodies
Enhancing Detection of Low-Abundance Proteins
9 tips for detecting phosphorylation events using a Western Blot
Western Blotting with Tissue Lysates
Immunohistochemistry introduction
Immunohistochemistry and Immunocytochemistry
Immunohistochemistry troubleshooter
Chromogenic and Fluorescent detection
Preparing paraffin-embedded and frozen samples for Immunohistochemistry

Western Blot Troubleshooting
Overview:
Western blotting is a commonly used laboratory technique for quantitative assessment of protein expression, but sometimes mistakes can happen. If you are able to isolate and iterate through the appropriate steps to find a solution you can quickly get back on track with your work.
In this article we outline a variety of common issues you may find while carrying out this technique and build on 4 major areas where errors typically arise - signal interference, lack of protein signal, faint bands and background noise.
Part one - Signal Interference
Why are the bands at lower molecular weight than expected?
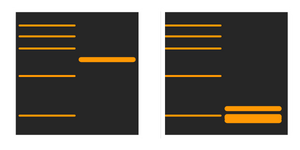
This issue can be caused by several things. One consideration is whether the target protein may be prone to cleavage or digested, which can result in signals at a lower molecular weight. Splice variants may also be present in the sample, or a different protein with similar epitope could have been detected by the antibody.
To counter these issues you can try using a fresh sample, kept on ice or in the freezer. In a fresh lysate from a fresher sample cellular enzymes are less likely to have broken down the proteins in the sample.
You could also try adding protease inhibitors to the lysis buffer, or trying an alternative primary antibody to ensure the right protein has been targetted.
Why are the bands slightly higher than expected and somewhat blurred?

Protein bands which are found at molecular weight signals, and which are higher than expected for the target protein, are most likely caused by protein glycosylation or post translational modifications across multiple amino acid residues on the protein of interest.
Phosphorlated, acetylated and methylated groups are common across a variety of proteins and their charge will contribute to the protein's migration through a gel.
We recommend checking the amino acid sequence of the protein in common databases - such as uniprot - to identify any modification sites, and if necessary apply appropriate enzymes to the lysate to remove modifications.
Why are the bands much higher than expected?
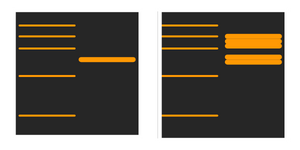
If the samples have not been fully reduced and denatured, protein-protein interactions, dimers and multimers might have occurred. Using a fresh loading buffer for the new samples will ensure that the gel runs more smoothly.
Why can I see multiple bands in different locations?
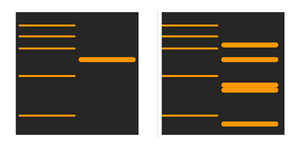
There are two major reasons for this to happen. Either the primary antibody concentration is too high, accompanied by cross-reactivity with similar epitopes, or the secondary antibody concentration is also too high, leading to merging of bands and non-specific binding.
Figuring out the appropriate dilution of either antibody will help take care of this issue. Switching antibodies may also help identify if there is cross-reactivity between the 2 antibodies.
Why are the bands too blurry?

If you have run the gel at a high voltage for a short period of time, the proteins may not run in a uniform manner, causing blurry bands.
Blurry bands can also occur if a mistake is made during the preparation of the running buffer, or if bubbles were trapped during the protein transfer stages.
This issue can be resolved by checking literature for the particular protein weight (different weights will have optimal results with different buffer and gel compositions, as well as casette loading times), preparing a new buffer and minimising human error.
Why do the binds look like a smile?
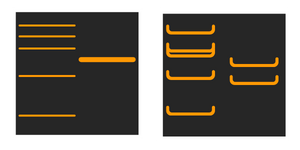
Smiling bands are caused when the proteins don't align correctly in the stacking gel as a result of high current. Typically the gel overheats and the proteins separate out of line.
Store your buffer in a fridge prior to use, and apply cool packs to the tank buffer zones, if possible. Run the gel at a lower current for longer, and optimise the electrophoresis conditions to flatten the bands.
It is also good practice to load the protein lanes slowly and as close to the bottom as possible (without coming into contact with the gel).
Why do I see white bands, when using ECL detection?

Too much loaded protein or too high antibody concentration can cause this. Check the protocol and optimise the concentration of the primary will remove help resolve this issue.
Part two: No bands:

Having no bands is a complex issue. The causes can be grouped in several categories: antibody, antigen, technique or buffer related.
Antibody related issues: Using the wrong secondary antibody:
Checking compatibility and redoing the experiment with the proper secondary antibody can ensure the appearance of bands. Furthermore, checking if the antibody is compatible with Western blotting and if correct concentrations are used before starting the experiment will ensure that no issues with antibodies will occur in the course of the Western blotting.
Antibodies need to be refrigerated at all times prior to the experiment, so the cold chain and their integrity are maintained.
Antigen related issues:
Usually, the lack of bands is caused by improper technique rather than a lack of antigen in the target tissue. To ensure no proteins are lost during the transfer to the membrane, Ponceau S stain can be used prior to the immunostaining as a verification.
Technique related issues:
Excessive washing between the different steps can remove a portion of the detection reagents, resulting in a poor outcome. Try using less aggressive techniques when washing and also shorten the duration and number of washes.
Buffer related issues:
Using fresh and sterile buffer is necessary, as any bacterial contamination will hamper the results.
Part three: Weak signal:

Changing the blocking agent and its concentration can help manage the signal interference. Also, ensuring that the peroxide and detection buffers used are fresh will help with avoiding contamination and the quality of the readings.
Antibody related issues:
Two of the most common issues resulting in faint bands are having insufficient antibody concentration or the antibody-antigen binding specificity is too low.
To take care of that, either optimizing the concentration alongside reducing the number and duration of washes and increasing the antibody concentration over the recommended amount are the first things to try.
Antigen related issues:
If the amount of sample loaded on the gel is too little or the stripping was too invasive, the signal obtained will be too weak.
To take care of these issues, checking the concentration and elevating it if needed is recommended, but it’s best to redo the blot, simply because the antigen might be damaged.
Technique related issues:
As usual, if the protocols are followed precisely and with great care, the experiment should run smoothly. But sometimes, confirming the protein transfer with a reverisible stain like Ponceau S before the immunostaining and optimizing the exposure times can be quite helpful.
Buffer related issues:
Non-fat dry milk has the tendency to mask some of the antigens. You can try swapping the blocking solution or instead to lower the percentage of milk in the solutions.
Part four: Background signal is high:

Antibody related issues:
If the primary or secondary antibodies’ concentrations are too high, or they bind to the blocking agent, high background can occur.
As before, following the recommended concentrations and optimizing them if they are not suitable for the specific experiment is the best option, accompanied by going for a different blocking solution if you are sure that the issue is caused by the binding.
Antigen related issues:
The most common issue is non-specific genomic DNA interactions in the sample. Usually, the addition of suitable DNAse to the lysis buffer and sonication of the lysates is enough.
Technique related issues:
Apart from the obvious issues you will always experience if you do not follow the protocol, the PVDF membrane can cause a higher background. If this is the case, try using a nitrocellulose one instead.
Furthermore, never let the membrane dry out, incubate it at too high of a temperature or overexpose the film. Most of the duration and temperature guidelines can be found in the protocols or product cards. Reading them before the experiment is always a good idea.
Buffer related issues:
With buffers, using fresh and sterile buffers and having adequate concentration can guarantee that you won’t experience any issues with them.
Part five: Patchy spots on the blot:

Antibody related issues:
Spinning the secondary antibody and filtering the aggregates out before the experiment should take care of any possible issues with it.
Technique related issues:
If any air bubbles are trapped in the membrane during the transfer, they can cause patchy spots to appear all over the blot.
During the incubation steps, uneven agitation can cause similar issues. Using a shaker or a rocker for those steps is advisable as it helps mitigate uneven agitation.
Lastly, all equipment and buffers used must be sterile and free of old pieces of gel or proteins to avoid any bacterial contamination or gel remnants sticking to the membrane.